Expression Information
In RNA sequencing (RNA-Seq), gene expression is measured by counting the number of reads that align to each gene, but raw counts need to be normalized for factors like gene length and sequencing depth. RPKM (Reads Per Kilobase of transcript per Million mapped reads) normalizes by adjusting for both gene length and total read count within a sample, making it useful for comparing gene expression within a single sample. TPM (Transcripts Per Million) also accounts for gene length but normalizes after adjusting for gene length, making it better for comparing expression across different samples. For cross-sample comparisons, methods like DESeq2 and EdgeR are preferred, as they use statistical models to correct for sequencing depth and other biases, providing more accurate results for differential gene expression analysis.

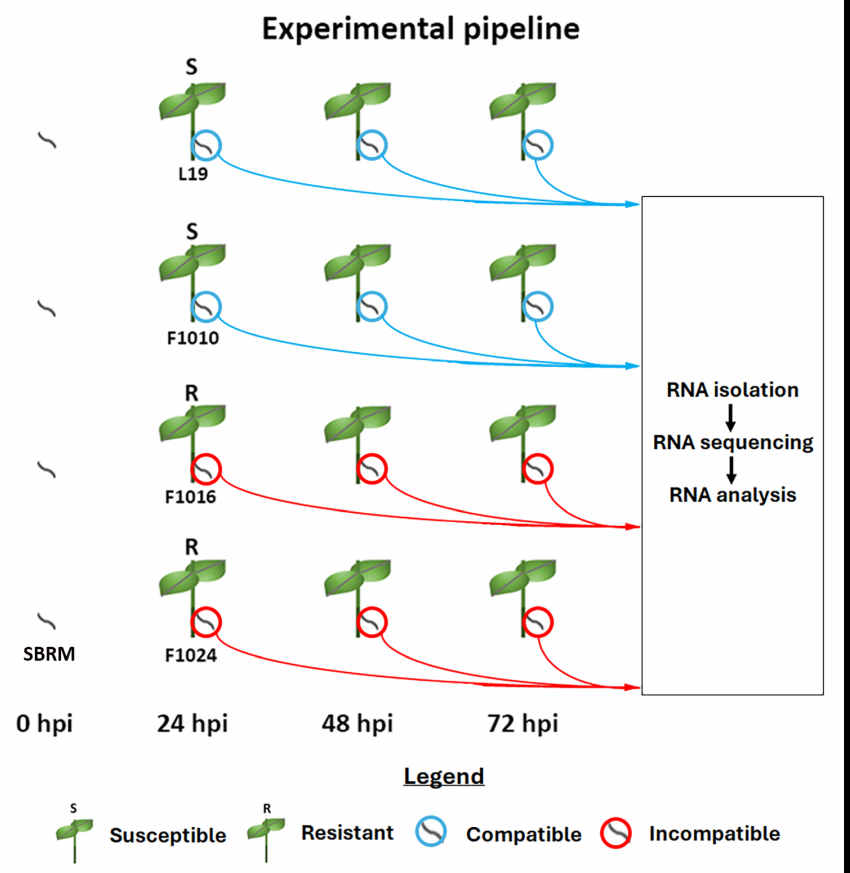
Figure 1. Experimental pipeline. The SBRM-susceptible L19 and F1010, and SBRM-resistant F1016, and F1024 B. vulgaris genotypes are shown. The respective compatible and incompatible SBRM are encircled by a blue or red ring. At t = 0 hpi, the SBRM were collected before any introduction to B. vulgaris. Thus, the SBRM are shown to not be closely associated with B. vulgaris. SBRM are subsequently shown to be in direct contact with B. vulgaris at the t = 24, 48, and 72 hpi time points. The samples were collected for transcriptomic study that involved RNA isolation, sequencing, and analysis.